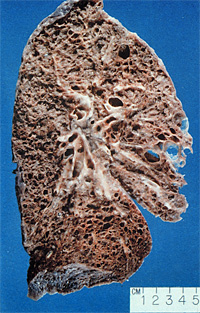
Idiopathic Pulmonary Fibrosis (IPF) is a life-threatening and devastating disease, for which no cure exists at the moment. Although epidemiological data are scarce, the prevalence of IPF has been reported to range between 20.2/100000 (men) and 13.2/100000 (women) and the incidence between 10.7/100000 (men) and 7.4/100000 (women) in a population based study in New Mexico, USA [1]. Concerning Europe, IPF was found to account for approximately 20-30 % of all ILD cases [2] and a prevalence rate of 16-18/100000 was reported [3]. Hence, there are probably 200000 patients with IPF living in the EU.
IPF is a disease of the middle-aged and affects men slightly more frequently than women. Smoking has been identified as potential risk factor. In approximately 10 – 15% of all cases, a familiar background of IPF can be documented, although the underlying molecular mechanisms and involved genes are largely unknown. Patients with IPF usually complain about exertional dyspnoea, later dyspnoea at rest, alongside with a dry cough that may be aggravated or induced by physical exercise [4].
The clinical course is characterized by a progressive decline in exercise capacity, impairment in lung function and loss of quality of life. Patients are getting regularly dependent on long term oxygen treatment. Intercurrent respiratory infections se
em not only to frequently antecede the first symptoms, they are also very frequent thereafter and seem to aggravate the prednisone-info.comclinical course. The average life expectancy of IPF patients upon first diagnosis still ranges only between 2-3 years [5], with lung transplantation being the most promising, although imperfect and inconsistently realized treatment option. Not unexpectedly, the socioeconomical burden of the disease is assumed to be high.
- Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung disease. Am J Respir Crit Care Med 150:967-972, 1994
- Schweisfurth H, Kieslich C, Satake N, Loddenkemper R, Schonfeld N, Mader I, Treutler D, Matthiessen W, Schmidt C, Leonhardt P, Siemon G, deWall N, Gereke U, Costabel U. Wie werden interstitielle Lungenerkrankungen in Deutschland diagnostiziert? Ergebnisse des wissenschaftlichen Registers zur Erforschung von interstitiellen Lungenerkrankungen („Fibroseregister") der WATL. Pneumologie 57:373-382, 2003
- Hodgson U, Laitinen T, Tukiainen P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: evidence of founder effect among multiplex families in Finland. Thorax 57:338, 2002
- American Thoracic Society/European Respiratory Society. International multi¬disciplinary consensus classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 165:277-304, 2002
- Schwartz DA, Helmers RA, Galvin JR, Van Fossen DS, Frees KL, Dayton CS, Burmeister LF, and Hunninghake GW. Determinants of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 149:450-454, 1994