The pathomechanism of IPF is yet not understood. Previously, it was anticipated that the disease reflects another inflammatory driven form of lung fibrosis. However, the frustrating experience in IPF is that steroids and immunosuppressants are of little help, if at all. A more recent and increasingly favored hypothesis puts the alveolar type II cell at the center of a unifying concept, according to which chronic epithelial damage is the underlying trigger mechanism for the development of lung fibrosis. In more waterblock.nldetail, misfolding or defective processing of proteins (e.g. surfactant protein C), altered lysosomal transport or processing or chronic DNA damage have already been proven to induce alveolar epithelial apoptosis. Hyperproliferation of these cells is induced to overcome the loss of epithelium and the factors released may – in a paracrine fashion - largely induce the uncontrolled proliferation of fibroblasts and the excessive deposition of extracellular matrix, mostly collagen.
a) What is the triggering event in IPF?
The alveolar epithelial type II (ATII) cell is thought to be at the center of a pathomechanistic concept for sporadic or familial IPF, but also for other DPLD, such as the Hermansky-Pudlak syndrome, amiodarone-induced pneumonitis, or irradiation-induced lung fibrosis. Both enhanced ATII cell apoptosis and hyperplasia have been reported in IPF specimen, ultimately inducing distorted epithelial-mesenchymal cross-talk, resulting in enhanced fibroblast activation and ECM synthesis.
Ultrastructural studies have revealed the existence of proliferative alveolar epithelial cells immediately adjacent to injured epithelial cells, suggesting that epithelial apoptosis and proliferation and hyperplasia occur simultaneously during the process of fibrosis. In detail, chronic endoplasmic reticulum (ER) or lysosomal stress have been reported to induce ATII cell apoptosis and thus set the stage for the development of lung fibrosis. In detail, mutations in the SFTPC (Surfactant Protein (SP)-C) and the TR/TERT (telomerase) genes in familial forms of idiopathic interstitial pneumonias (mostly IPF and NSIP) provided initial evidence that apoptosis of ATII cells may represent an important pathogenetic trigger event.
In parallel, activation of ER stress pathway components including endoplasmic reticulum to nucleus signaling 1 (ERN1) (by proof of X-box binding protein (XBP) 1 splicing) and activating transcription factor (ATF) 6 was observed in IPF and NSIP, and found to result in a persistent and overwhelming ER stress response and induction of epithelial apoptosis via DNA-damage-inducible transcript (DDIT) 3. Also, respiratory infections represent a common phenomenon in IPF, which not only seem to frequently antecede the clinical appearance of the disease, but also to accelerate its clinical course. Consistent with these observations, bacterial, and – even more compelling – viral infections can induce severe ER-stress. Thus, an intriguing and unifying concept for sporadic and familial IPF would consist of a genetic predisposition to an epithelial injury, a modifying environmental stimulus and a common downstream pathway resulting in fibrosis based on ER stress (or DNA damage) induced ATII cell apoptosis.
Other diseases resulting in chronic ATII cell injury, such as Hermansky Pudlak Syndrome, amiodarone- or irradiation-induced lung fibrosis, might similarly result in epithelial apoptosis and subsequent fibrosis, and could thus be integrated into this concept.
b) What are the mediators of distorted epithelial-mesenchymal interactions in lung fibrosis?
Balanced epithelial-mesenchymal interactions are of the utmost importance for proper lung development, in particular for regular definition of a proximal-distal axis and dichotomous branching. In the adult lung, mesenchymal-epithelial interactions warrant proper lung function and are a prerequisite for the maintenance of the trophic alveolar unit, but impaired epithelial-mesenchymal crosstalk between ATII cells and subepithelial fibroblasts, as well as dysregulated precursor cell recruitment, have recently been shown to contribute to the pathobiology of IPF.
It has been proposed that the ATII cell, by action of cyclooxygenase 2, releases PGE2, which then binds to the EP-2 receptor on fibroblasts, increases cAMP levels in the fibroblast and thereby inhibits the proliferation and transactivation of this cell. In addition, several growth factors are released by the ATII cell that control the fibroblast phenotype, such as members of the Wnt, BMP, or TGF-β superfamilies. In particular, enhanced secretion and/or activity of Wnt and TGF-β superfamily members have been documented in IPF. The fibroblast itself is a rich source of FGF-7, FGF-10, and HGF. HGF will be released by fibroblasts in dependency of cAMP levels and must be activated by extracellular serine proteases such as the HGF activator. FGF-7, FGF-10, as well as HGF are known to exert a marked influence on ATII cell proliferation, migration and survival, and at least HGF has been shown to be released to a much weaker extent from IPF fibroblasts as compared to fibroblasts from healthy lungs.
Thus, loss of regenerative capacity of the resident ATII cell population due to loss of FGF-7, FGF-10, or HGF may contribute to the pathogenesis of IPF.
c) What is the origin of activated (myo-)fibroblasts in IPF?
While the initial injury in IPF is most likely affecting the ATII cell (see above), it is well accepted that the interstitial fibroblast / activated myofibroblast represents the key effector cell responsible for the increased ECM deposition that is characteristic for this disease. The number of smooth muscle actin-positive, activated (myo)fibroblasts is significantly increased in multiple forms of pulmonary fibrosis including IPF, but their origin remains to be elucidated.
Currently, three major theories attempt to explain this hallmark of maladaptive cell activation. It has been demonstrated that resident pulmonary fibroblasts proliferate in response to fibrogenic cytokines and growth factors (see below), thereby increasing the local fibroblast pool via local fibroproliferation. In addition, several recent studies have shown that bone marrow-derived circulating fibrocytes traffic to the lung during experimental lung fibrosis, and serve as progenitors for interstitial fibroblasts.
In particular, collagen I-positive fibrocytes have been shown to traffic to injured lungs in a chemokine-dependent fashion, integrate into the lung ECM, and contribute to enhanced collagen synthesis in fibrosis.
Third, it was recently proposed that ATII cells are capable of undergoing the process of epithelial-to-mesenchymal transition (EMT), the phenotypic, reversible switching of epithelial to fibroblast-like cells, which is initiated by an alteration of the transcriptional and proteomic profile of AT2 cells. The orchestrated series of events initiating EMT include remodeling of epithelial cell-cell and cell-matrix adhesion contacts, reorganization of the actin cytoskeleton, and induction of mesenchymal gene expression.
EMT is a highly controlled process initially discovered and described in embryonic development and morphogenesis. In addition, EMT has gained wide recognition as a mechanism that facilitates cancer progression and metastasis, as well as the development of chronic degenerative fibrotic disorders of the kidney, liver, and lung. Transforming growth factor (TGF)-β is a main inducer and regulator of EMT in multiple organ systems.
d) What are the major signaling pathways underlying matrix remodelling in the lung?
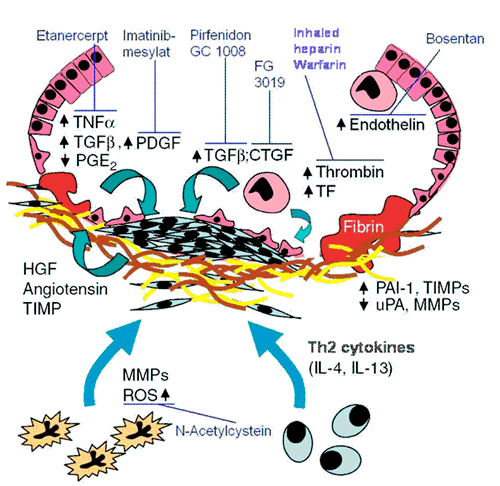
As a kind of primary wound healing response, activation of the coagulation cascade and suppression of the fibrinolysis system has been observed in patients with IPF, and the cellular origin of these coagulation factors (alveolar macrophages and alveolar type II cells) has been disclosed by UGLC members. Analysis of bronchoalveolar lavage fluids (BALF) revealed substantial activation of the extrinsic coagulation pathway (tissue factor [TF]; FVII), alongside with pronounced suppression of antithrombotic (activated protein C) or fibrinolytic (Plasminogen Activator Inhibitor [PAI]-1) activities. These changes promote alveolar and interstitial fibrin deposition, forming a provisional matrix and thereby substantially contributing to lung fibrosis. Moreover, several procoagulant serine proteases such as TF/FVII, factor X and thrombin induce fibrotic events via the Protease activated receptor (PAR)-1 and PAR-2. In response to the activation of this G-protein coupled receptor, increased ECM production and secretion and induction of profibrotic growth factors such as TGF-ß and PDGF can be observed. Vic versa, the urokinase system has repeatedly been shown to exert strong antifibrotic activity, most likely due to the activation of HGF and the removal of fibrin and ECM. Persistent suppression of urokinase by PAI-1 overexpression, as seen in IPF patients and in animal models of lung fibrosis, would thus contribute to the development of lung fibrosis. As also shown by UGLC members and other groups, alveolar deposition or overexpression of urokinase, knock out of PAI-1 or inhibition of the procoagulant pathways by heparin, factor Xa antagonists, direct thrombin inhibitors or activated protein C all resulted in a substantial suppression of the fibrotic response in the bleomycin model of lung fibrosis, whereas knock out of urokinase or alveolar PAI-1 overexpression induced the opposite.
With respect to scar formation as aberrant alveolar/interstitial wound healing response, there is currently no doubt that the Transforming Growth Factor (TGF)-β family represents the pivotal mediator system. In vitro, TGF-β induces fibroblast chemotaxis, proliferation and transdifferentiation into myofibroblasts, and it largely promotes the production and secretion of extracellular matrix compounds, mainly collagen. Application of TGF-β encoding adenoviral vectors to the distal lung induces a progressive and severe lung fibrosis. Likewise, application of these vectors to the pleural space induces pleural fibrosis and subpleural lung fibrosis as seen in IPF. Increased TGF-β signaling is also observed in other animal models of lung fibrosis, such as the bleomycin model, where collagen deposition is reduced by TGF-β antibodies and soluble TGF-β receptors. In lungs of IPF patients, increased expression of TGF-β has been observed in close proximity to areas of increased ECM deposition. Apart from TGF-β, there are also other growth factors such as PDGF (platelet-derived growth factor), CTGF (connective tissue growth factor), members of the Wnt pathway, or IGF-I (insulin-like growth factor I) and endothelin, which may significantly contribute to the pathogenetic sequelae of IPF.
Apart from the proliferation of fibroblasts, the excessive deposition of matrix is a key feature of IPF and, most likely, is the result of excessive production of ECM compounds and a local imbalance between the matrix metalloproteinases (MMP) and their inhibitors (TIMP). In general, increased TIMP expression and virtual absence of the collagen I specific MMP-1 has been observed in the lungs of IPF patients, thus contributing to collagen deposition. In view of the coexistence of fibrotic scars and honeycomb cysts in the lungs of IPF subjects, it is yet not settled, if a spatial disarrangement of the collagenases (largely MMP-1) and the TIMPs may be the primary reason for the development of this structural heterogeneity. In contrast, the two gelatinases MMP 2 und 9, known for their ability to destruct the basement membrane and thus to impair epithelial regeneration, were found to be increased in lung fibrosis. Finally, matrilysin (MMP-7) was also found to be highly upregulated in IPF and MMP-7 knock out mice were protected from the bleomycin induced lung fibrosis, thus supporting an important role of this MMP.
Treatment of lung fibrosis - translational approaches
In striking contrast to the field of Pulmonary Arterial Hypertension, a disease previously characterized by a similarly poor outcome, the therapeutic approach to patients with IPF has not changed dramatically over the last 10 years. Apart from standard care, including long-term oxygen treatment, aggressive and early treatment of respiratory infections, and early listing for lung transplantation if suitable, all recently finished phase II/III trials exhibited unsatisfactory results. To this end, interferon-γ definitely proved ineffective in IPF. One larger trial in IPF with a significant improvement in the primary study endpoint, albeit with a high drop out rate (1/3 of all patients), was the IFIGENIA trial, in which n-acetyl cysteine was tested against placebo and was found out to attenuate the loss of lung function. Additionally, Pirfenidone (targeting the TGF-β pathway), recently approved in the EU, provides hope for the slowing of disease progression. Overall, an encouraging increase in clinical trials in the field of IPF can be observed. Most of these studies are addressing secondary processes forwarding fibrosis per se.
An overview is given in the figure outlined below.